2.2 Alteraciones Farmacodinámicas Relacionadas Con La Resistencia Al Tratamiento Hormonal
-
Papel de los estrógenos intratumorales. En las células tumorales se suelen alcanzar niveles de estrógenos más elevados que en el suero debido a que: 1) el tejido mamario los concentra en sus compartimientos intra y extracelulares y 2) las células sintetizan estrógenos a través de rutas similares a las presentes en los tejidos adiposos periféricos. Esto sugiere que aquellos mecanismos o fuentes endógenas (p. ej., ovarios o tejido adiposo) o exógenas (p.ej., estrógenos ambientales o anticonceptivos) que incrementen los niveles séricos de estrógenos, y consecuentemente contribuyan a elevar los niveles intratumorales, pueden modificar la respuesta celular a los antiestrógenos. La competición de los estrógenos por el RE impide el antagonismo eficaz de los antiestrógenos sobre el receptor y de esta forma contribuyen a desarrollar un fenotipo de resistencia endocrina. Por ejemplo, la presencia de ovarios funcionales disminuye la respuesta de los antiestrógenos. Esto se puede explicar por las acciones de los antiestrógenos a nivel de los mecanismos de regulación de la liberación de gonadotropinas hipofisarias. Cabe esperar que si el TAM bloquea el RE en el hipotálamo y/o la hipófisis, los estrógenos circulantes no inhibirán la liberación de gonadotropinas y, en última instancia, habría una situación de hiperestimulación ovárica. Esto podría explicar cómo el TAM incrementa los niveles circulantes de estrógenos lo cual contribuiría a un aumento de sus niveles intratumorales y, como explicamos anteriormente, a un fenotipo de resistencia hormonal. Otro mecanismo, al menos hipotético, que podría contribuir a un fenotipo de resistencia endocrino está relacionado con la sobreexpresión de RE en los tumores lo cual condicionaría un estado de hipersensibilidad celular a los estrógenos. Por último, se estima que, en ausencia de sobreexpresión del RE, sólo está ocupado un 25% del total de los receptores lo cual sugiere que muchos tumores de mama existen en un ambiente estrogénico relativamente débil. Esto podría explicar cómo concentraciones subóptimas de estrógenos, como las detectadas en algunos tumores, puedan soportar la hipótesis de que diversas fuentes estrogénicas medioambientales, aparentemente inocuas, estén asociadas con el desarrollo de cáncer de mama.
-
Actividades mixtas agonista/antagonista de los SERMS. Las consecuencias biológicas derivadas de la ocupación del RE por un antiestrógeno y, por lo tanto, sus acciones estrogénica o antiestrogénica dependen de, al menos, los siguientes factores: 1) el contexto celular, 2) el tipo de RE y 3) la estructura del ligando. El TAM es un excelente ejemplo de agonista/antagonista mixto que ilustra estos aspectos.
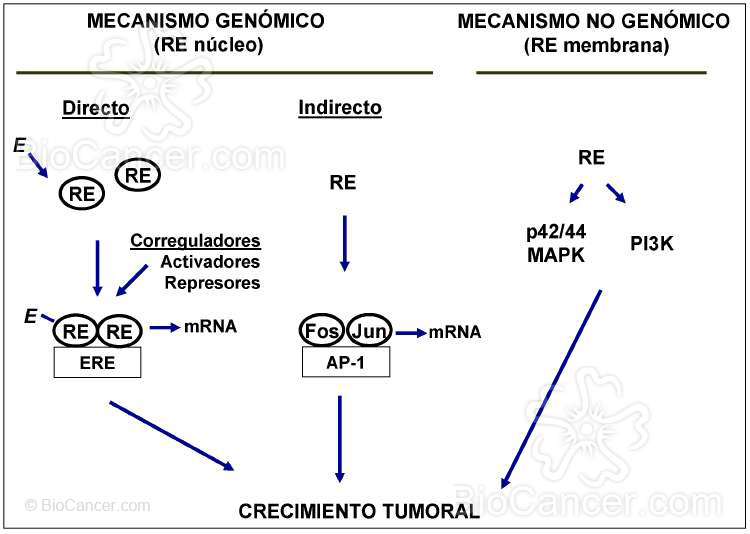
Figura 2. Funciones del RE. Mecanismos genómico: después de la unión a su ligando (E), el RE nuclear
activa la transcripción a través de la unión directa con sus elementos de respuesta en los genes dianas (modo
clásico) o bien controlando a otros factores de transcripción c om o el complejo AP-1 (modo no clásico).
Mecanismo no genómico: el RE de la membrana, a través de la interacción directa con diferentes
interemediarios de señalización intracelular próximos en la membrana, pude dar lugar a la inducción rápida
de cinasas claves de receptores para factores de crecimiento como p42/44 MAPK o PI3K.
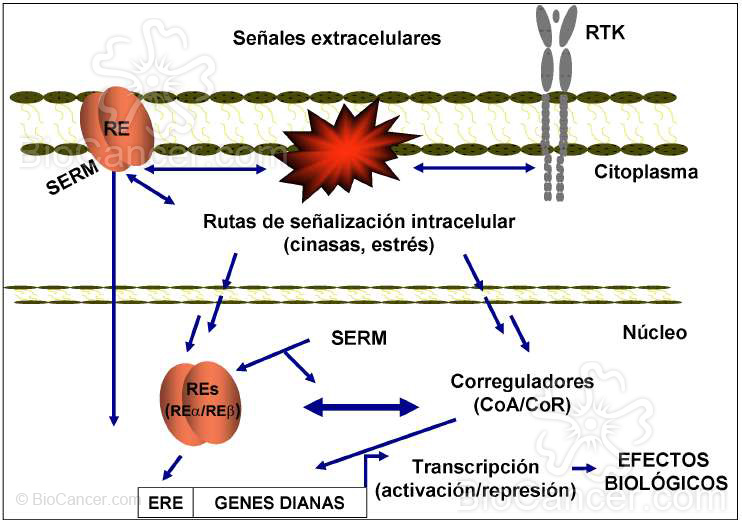
Figura 3. La acción selec tiva de los SERMs en una determinada célula o tejido es la resultante de la combinación
de diferentes efectos dependientes de un delicado balance entre múltiples factores interconectados que actúan de
forma concertada para modular las acciones del RE. Estos incluyen los niveles y ratios de los diferentes subtipos
de RE, proteínas correguladoras de la transcripción, señales extracelulares y rec eptores para factores de crecimiento
o cinasas o la naturaleza de los promotores de los genes dianas. Las acciones del RE ocurren predominantemente
en el núcleo pero sus efectos en la membrana también pueden contribuir a los actividades de los SERMs
Por ejemplo, el TAM produce una respuesta estrogénica en el hueso y endometrio y antiestrogénica en la mama. El patrón de respuesta inverso podría inducir el crecimiento del tumor y el riesgo de sufrir fracturas. Afortunadamente, durante los últimos años se han aportado excitantes datos experimentales sobre cómo los estrógenos y el RE activan el crecimiento de las células de cáncer de mama y cómo los SERMs pueden modificar la actividad del RE (Fig. 2 y 3). En este contexto, uno de los objetivos de la terapia antineoplásica en el cáncer de mama es encontrar SERMs que mantengan los efectos beneficiosos del TAM (antagonismo en el cáncer de mama) pero sin un mayor riesgo de inducir tumores (agonista en el carcinoma de endometrio) (11).
-
Mecanismos de resistencia dependientes del RE. La función ago ni st a de l os S ERMs se relaciona, fundamentalmente, con el dominio funcional AF-1 del RE y la modulación de este dominio activador es crítica en la resistencia al TAM. Es muy interesante resaltar que para algunos SERMs, incluido el TAM, se ha demostrado una actividad agonista preferente sobre el RE . Esto sugiere que los dos REs señalizan por mecanismos diferentes, aún no muy bien definidos, como pueden ser a través de la interacción con otros factores de transcripción y sin que necesariamente implique una interacción directa del RE con el ADN (12-15) y por mecanismos que implican una interacción directa con el ADN. Por otra parte, no debemos olvidar que tanto los estrógenos como los SERMs también ejercen acciones no-genómicas (Fig. 2) que pueden contribuir al fenotipo de resistencia (9). El significado biológico de este modo de acción en el cáncer de mama no ha sido del todo clarificado. Mayor información se tiene sobre las bases moleculares que parecen explicar la participación específica del RE en el fenómeno de resistencia hormonal y que son, al menos, las siguientes: a) cambios genéticos en el RE, b) su relación con las proteínas coreguladoras de la transcripción (correpresores, coactivadores) o c) sus interacciones con otras rutas de señalización intracelular.
a.
cambios genéticos en el RE: mutaciones. Los fenómenos de amplificación y/o reorganización del gen RE so raros en el cáncer de mama. Sin embargo, sí se han identificado formas de maduración alternativa del ARNm que carecen de exones específicos y que podrían codificar para formas truncadas del RE constitutivamente activas y funcionar como un mecanismo de resistencia a la terapia endocrina.
b.
proteínas coreguladores de la transcripción. En general, las proteínas correpresoras sólo son reclutadas cuando el RE está unido al antagonista. Por lo tanto, en presencia de los SERMs con agonismo / antagonismo mixto, los complejos de correpresores pueden ser reclutados por el RE pero con el resultado de una represión incompleta de la transcripción. Por el contrario, las proteínas coactivadoras se unirían al RE cuando éste es ocupado por agonistas. Ahora bien, en relación con el fenotipo de resistencia hormonal parece ser que los niveles de expresión y/o actividad de las proteínas correguladoras de la transcripción y, por lo tanto, todos aquellos mecanismos que controlan su regulación, juegan un importante papel. Estos mecanismos de regulación no sólo controlan el ratio coactivadores/correpresores sino que pueden ser diferentes entre los diferentes tejidos dianas del RE. Esto indica que la diferencia relativa de coactivadores y correpresores, bien a nivel de expresión y/o actividad, puede modular no sólo las acciones del RE sino también la relación agonista/antagonista de los SERMs. Este modelo puede explicar las acciones específicas de tejido de los SERMs y la resistencia hormonal. Por ejemplo, la sobreexpresión y/o mutación de algunas proteínas coactivadoras (p.ej., AIB1) contribuye a generar resistencia tumoral contra el tratamiento endocrino (16). Estos datos sugieren que una valoración de los niveles de expresión y/o actividad de coactivadores/correpresores en el cáncer de mama podría ser esencial para predecir el pronóstico y la respuesta a la terapia en un paciente determinado.
c.
interacciones con otras rutas de señalización intracelular.Las interacciones existentes entre el RE y las rutas de señalización dependientes de receptores de la familia EGFR/HER2 e IGF-I contribuyen a la resistencia endocrina y operan en ambas direcciones.
1.
el RE regula la expresión y/o actividad de elementos de otras rutas de señalización. En el cáncer de mama, los estrógenos y el RE inducen la expresión y/o activación de diferentes proteínas involucradas en las vías de señalización reguladas por EGF/HER2 e IGF-I. Por ejemplo, los estrógenos inducen la expresión del receptor para IGF-I o factores de crecimiento autocrinos (p.ej., TGF e IGF-II). Además, a través de sus acciones sobre proteínas de membrana, el RE, dependiendo de la interacción con el estrógeno, puede activar directamente a los receptores de membrana y, consecuentemente, a sus respectivas cascadas de fosforilación y moléculas de señalización (9, 17). Un incremento anormal de la señalización de vías dependientes de factores de crecimiento parece ser responsable de la pérdida de una dependencia estrogénica dando lugar a tumores resistentes a antiestrógenos. Por ejemplo, la resistencia adquirida en células MCF-7 in vitro, después de un tratamiento prolongado con TAM, está asociada con niveles incrementados de EGFR y actividad MAPK.
2.
Modificaciones post-transcripcionales del RE: fosforilaciones. A pesar de que la actividad del RE ha sido clásicamente descrita como dependiente de la interacción con ligando, diferentes factores de crecimiento (p.ej., EGF, IGF-I) pueden estimularla en ausencia de ligando. Pero, no sólo el RE sino también otros elementos de su cascada de señalización pueden ser regulados por fosforilación (9, 17). Los correguladores de la transcripción puede ser fosforilados en múltiples sitios y por diferentes cinasas (p. ej., p42/44MAPK, AKT, JNK, p38 MAPKs, PKA). Esto sugiere que si la célula expresa niveles elevados tanto de coactivadores como de cinasas se podrían incrementar, marcadamente, las propiedades agonistas del antiestrógeno. Parece ser que la fosforilación potencia la localización nuclear de los coactivadores y su interacción con el RE y puede, directamente, incrementar sus propias actividades acetiltransferasa o estimular sus habilidades para reclutar otros coactivadores transcripcionales o integradores al complejo receptorial. Tal como ocurre con los coactivadores, diferentes vías de señalización intracelular también pueden atenuar las actividades de los correpresores sobre el RE. La estimulación de la actividad RE por mecanismos de fosforilación está comúnmente mediada por las siguientes rutas:
-
ERK1/2.
Fosforila a la serina 118 del AF1 dando lugar a una transactivación del RE independiente del ligando (18). La propia unión del ligando al RE induce la fosforilación de la serina 118 (19) a través de CDK7, una cinasa dependiente de ciclina que está asociada con el factor de transcripción basal TFIIH (20). Por lo tanto, un incremento de la actividad cinasa (p.ej., ERK) podría ocasionar resistencia a la terapia hormonal. En este sentido, se ha demostrado que la expresión y/o a actividad de ERK1/2 están incrementadas tanto en modelos celulares de cáncer de mama con resistencia hormonal (21) como en cánceres de mama humanos en relación con el tejido de mama sano (22). Un incremento de la actividad ERK1/2 se correlaciona con una peor respuesta al tratamiento hormonal y con una supervivencia menor (23).
-
PI3K- AKT.
Otra vía de señalización que parece tener un papel importante en la actividad del RE es la dependiente de PI3K. Uno de las proteínas reguladas por PI3K es la serina/treonina cinasa AKT (PKB) (24). Su activación promueve la proliferación y la respuesta antiapoptótica. La fosforilación del RE por AKT se produce en la serina 167 y conduce a una activación del RE independiente del ligando (25) (26). A favor de la importancia de AKT está que tanto la expresión como la actividad de todos los miembros de esta familia de cinasas están incrementados en los cánceres de mama y, frecuentemente, se debe a una amplificación de su gen (27). Por lo tanto, la actividad alterada de la vía PI3K-AKT podría ser importante como mecanismo de resistencia a la terapia endocrina.
3.
El eje Hormona de Crecimiento-IGF-I. El eje hormona de crecimiento (GH)-factor de crecimiento similar a insulina tipo I (IGF-I) juega un papel determinante tanto en el desarrollo de la mama normal como del cáncer de mama. Además, parece estar involucrado en el desarrollo de resistencia al tratamiento con TAM o con Herceptin.
La relación existente entre los estrógenos y el eje GH-IGF-I es muy estrecha y ocurre a diferentes niveles. Por ejemplo, el TAM bloquea la proliferación de células de cáncer de mama in vitro mediada por IGF-I (28) y suprime los niveles séricos de IGF-I in vivo. El efecto in vivo, es el resultado de diferentes acciones: a) inhibición de la secreción de GH; b) inhibición de la expresión de IGF-I en el tejido mamario, y c) inducción de IGFBP-3 que reduce la biodisponibilidad del IGF-I. El estradiol incrementa la biodisponibilidad del IGF-I porque inhibe la síntesis de IGFBP-3 e incrementa la de catepsina D, una proteasa de IGFBP-3 (29). Esta interacción tan estrecha se refleja a nivel funcional, observándose un pronunciado sinergismo de los estrógenos y de IGF-I sobre la proliferación de diferentes líneas celulares de cáncer de mama (30). Por otra parte, tanto en el tejido mamario normal (31) como en el cáncer de mama RE-positivo (32), el estradiol potencia la expresión de IGF-I dependiente de GH. La interacción del RE con el eje GH-IGF-I, es recíproca ya que el IGF-I puede regular la actividad del RE y la síntesis de proteínas dependientes de RE por mecanismos independientes del estrógeno. Finalmente, se ha descrito que IGF-I afecta directamente el metabolismo de los estrógenos en células de cáncer de mama modulando la biodisponibilidad de los mismos. Estas interacciones contribuyen al que el eje GH-IGF-I se implique en el desarrollo un fenotipo de la resistencia hormonal en el cáncer de mama (33). Por lo tanto, el control de las señales dependientes de IGF-I puede ser una buena aproximación para superar la resistencia de la célula cancerígena al tratamiento hormonal. El IGF-I también puede interferir con las acciones antiproliferativas de Herceptin. Esto está asociado a un incremento de la señalización a través del receptor de IGF-I. A este mecanismo hay que añadir que los receptores para IGF-I y ErbB2 pueden heterodimerizar dando lugar a un receptor sin capacidad para unir Herceptin (34).
El importante papel que juega el eje GH-IGF-I en el desarrollo y comportamiento del cáncer de mama así como su estrecha interacción con los estrógenos permite predecir importantes aplicaciones clínicas. Por ejemplo, los niveles séricos de IGF-I podrían ser un determinante importante en la reaparición de cáncer de mama y su determinación serviría para valorar el riesgo de padecer cáncer de mama.
-
Mecanismos de resistencia independientes del RE. Como hemos comentado anteriormente, se han identificado diferentes dianas para los antiestrógenos que son diferentes del RE. Estas se conocen como dianas no-genómicas porque no requieren la interacción del antiestrógeno con el RE y/o no afectan directamente las actividades transcripcionales de éste. Estas dianas han suscitado una considerable atención, ya que las acciones no-genómicas de los antiestrógenos (independientes del RE) pueden ser también relevantes en relación con su mecanismo de acción genómico (dependiente del RE). Como veremos a continuación, aunque las interacciones iniciales sean independientes del RE, las consecuencias derivadas de las mismas podrían afectar la expresión y/o actividades del RE.
a)
a) Antiestrógenos y estrés oxidativo. La generación de estrés oxidativo tiene efectos significativos sobre la regulación de diferentes genes (35) y puede, además, alterar substancialmente el contexto celular de las células afectadas. La capacidad de las especies reactivas de oxígeno de regular la expresión de genes depende de múltiples factores. Por ejemplo, depende de la existencia de un elemento de respuesta a agentes electrofílicos en el promotor de algunos genes sensibles a cambios en el estado de oxido-reducción de la célula (15).
El TAM, al igual que los estrógenos, es antioxidante, fundamentalmente, debido a su capacidad de influenciar las propiedades de la membrana. Sin embargo, existen evidencias claras de que el TAM, aparte de sus propiedades antioxidantes, induce una respuesta de estrés oxidativo en determinadas células. Esta aparente paradoja tiene ver con la capacidad de la célula para metabolizar al antiestrógeno y con la generación de metabolitos que son potencialmente proxidantes.
b)
Antiestr óg enos y pertur b ac iones en la memb r ana. Muchos antiestrógenos son lipofílicos y particionan, predominantemente, dentro de los dominios hidrofóbicos de las membranas celulares. Esto, es muy probable, que afecte las propiedades fisicoquímicas de los dominios de la membrana donde particiona el antiestrógeno y, consecuentemente, podría alterar las funciones de proteínas vecinas o adyacentes que son dependientes de las propiedades del ambiente lipídico para llevar a cabo su actividad. Entre estas, se incluyen receptores para factores de crecimiento, al RE y otras moléculas de señalización asociadas a la membrana (p.ej., proteínas G, PKC, fosfoinositoles, etc.) y que participan significativamente en fenómenos de proliferación o desarrollo tumoral. Por ejemplo, el TAM inhibe la actividad de PKC y de calmodulina lo cual contribuye a sus acciones antiproliferativas. Esto se explica en la medida en que PKC media la actividad mitogénica de determinados oncogenes y factores de crecimiento, y su actividad suele ser mayor en células neoplásicas de mama cuando se compara con tejido normal. Si las actividades de estas proteínas son factores limitantes para el desarrollo tumoral, cualquier inhibición significativa de su actividad puede ser suficiente para reducir la proliferación celular. Por lo tanto, perturbaciones bien de su nivel de expresión o de sensibilidad a la inhibición por TAM podrían contribuir al un desarrollo de un fenotipo de resistencia hormonal.
c)
Antiestrógenos y efectos no-genómicos. Los efectos no-genómicos de los antiestrógenos están normalmente regulados por mecanismos independientes del RE. Por lo tanto, se puede pensar que para conseguir una inducción total de de la respuesta celular a los antiestrógenos es necesaria la interacción entre los eventos mediados por el RE y los no-genómicos. Por otra parte, la respuesta de una célula al RE está condicionada por el contexto celular y, en este sentido, los mecanismos no-genómicos pueden contribuir marcadamente. Por ejemplo, determinados componentes de la cascada de señalización dependiente de antiestrógenos pueden ser expresados o reprimidos. Esto podría suceder por perturbaciones en las actividades de proteínas intracelulares claves en los mecanismos de señalización como PKC, calmodulina o diferentes factores asociados con la inducción de una respuesta de estrés oxidativo. Por ejemplo, el estrés celular está frecuentemente acompañado por cambios en la expresión de factores moduladores de la apoptosis como AP-1 o NFkB.
-
Alteraciones en el sistema inmune. Las actividades inmunosupresoras de los estrógenos se conocen desde hace muchos años y, en la actualidad, existen múltiples evidencias que demuestran la capacidad de los antiestrógenos para influir sobre diferentes aspectos de la inmunidad (9). Algunos de estos efectos están mediados por el RE mientras que otros reflejan perturbaciones en las actividades de dianas independientes del RE. Por lo tanto, las actividades inmunomoduladoras de los antiestrógenos pueden, potencialmente, contribuir no sólo a sus diferentes mecanismos de acción sino también al fenotipo de resistencia endocrina.