5.1 Influencia De La Actividad Cdk En La Inestabilidad Genómica
5.1 Influencia De La Actividad Cdk En La Inestabilidad Genómica
Mutaciones genéticas pueden permitir la sobreexpresión de ciclinas y CDKs. Asimismo, los inhibidores de CDK se pueden encontrar a menudo mutados, eliminados o silenciados en tumores, provocando la pérdida de funcionalidad de estos elementos en células cancerígenas, permitiendo su desarrollo y proliferación descontrolada (34,35,36).
Por otra parte, durante la tumorogénesis las células neoplásicas adquieren una serie de mutaciones genéticas que, al contrario de las células normales, les permiten escapar de la inhibición del ciclo celular y de la apoptosis, confiriéndoles una ventaja proliferativa adicional31 además de una capacidad invasiva que las células normales no poseen. Esta capacidad proliferativa puede deberse a la pérdida de puntos de control (checkpoints) en el ciclo celular, a la inactivación de los CDKIs, a la sobreexpresión de ciclinas y CDKs o a una combinación de todos estos factores (33).
Un claro ejemplo de esto es el gen que codifica a p16, un inhibidor del tipo INK4 especialmente sensible al silenciamiento epigenético, inducido por hipermetilación de su región promotora. Esto da lugar a la inhibición de la transcripción y pérdida de la expresión del gen, dando como resultado una proliferación descontrolada. Por consiguiente, la pérdida de la función de p16 se ha asociado a multitud de malignizaciones incluyendo melanoma, pulmón, pecho, y tumores colorrectales (33,37). Del mismo modo, la sobreexpresión de ciclina D1 se ha asociado con el desarrollo y progreso de cáncer de mama (33,38).
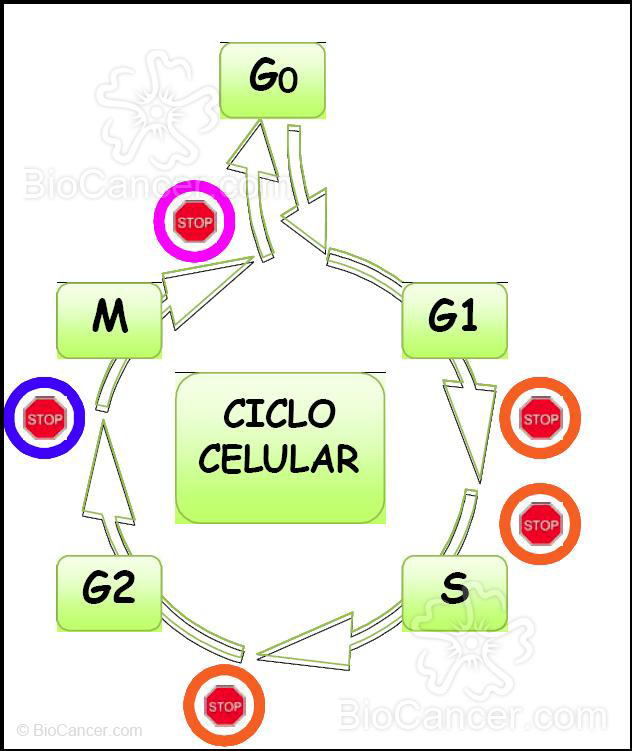
Figura 8. En el ciclo celular existen diversos puntos de control (checkpoints) que verifican el desarrollo del mismo en sus diferentes fases. Estos puntos verifican la correcta finalización de cada etapa del ciclo antes de dejar que comience la siguiente. Se pueden englobar en tres tipos diferentes según la función reguladora que ejerzan: puntos de control de daño al ADN (naranja), puntos de control de ADN no replicado (azul) y punto de control de la separación cromosómica (violeta).
A pesar de que la inestabilidad genómica es un rasgo común en tumores humanos, los mecanismos por los que ésta se produce no son muy conocidos31. Sin embargo, evidencias recientes sugieren que el daño al ADN podría estar extendido en estados pre-neoplásicos tempranos, siendo la desregulación de la actividad CDK una de las causas que lo provocan (31).
Asimismo, una actividad CDK aumentada podría reducir los controles en la replicación del ADN, conducir esta replicación, o mediar la sobreexpresión de las proteínas checkpoint, induciendo retrasos letales en el ciclo celular (31). A la inversa, la inhibición de la actividad CDK podría comprometer la eficiencia de la replicación, la expresión de las proteínas checkpoint, o la activación de las proteínas reparadoras de ADN31. Estas funciones vitales señalan el impacto de la actividad CDK en la estabilidad del genoma (31).
La desregulación de la actividad CDK en cáncer ha hecho de ésta una diana para el desarrollo de pequeñas moléculas inhibidoras de CDKs para el tratamiento de esta enfermedad. Sin embargo, esta estrategia terapéutica se ha visto complicada por dos factores:
Además, actualmente existen evidencias de que las CDKs, consideradas esenciales para el desarrollo del ciclo celular, podrían no ser realmente imprescindibles. Un ejemplo de esto es CDK2 (16,34,39). Estudios con ratones knock-out sugieren que esta quinasa no es esencial para la división mitótica de la célula (34,39). La inhibición de CDK2 demuestra que ésta no es indispensable para la progresión celular en cáncer y que por tanto no es una diana terapéutica adecuada (34,39). Una explicación de estos resultados es la existencia de una redundancia de CDKs en la célula, de manera que cuando CDK2 no esta disponible, la ciclina E podría combinarse con otra CDK (31,34).
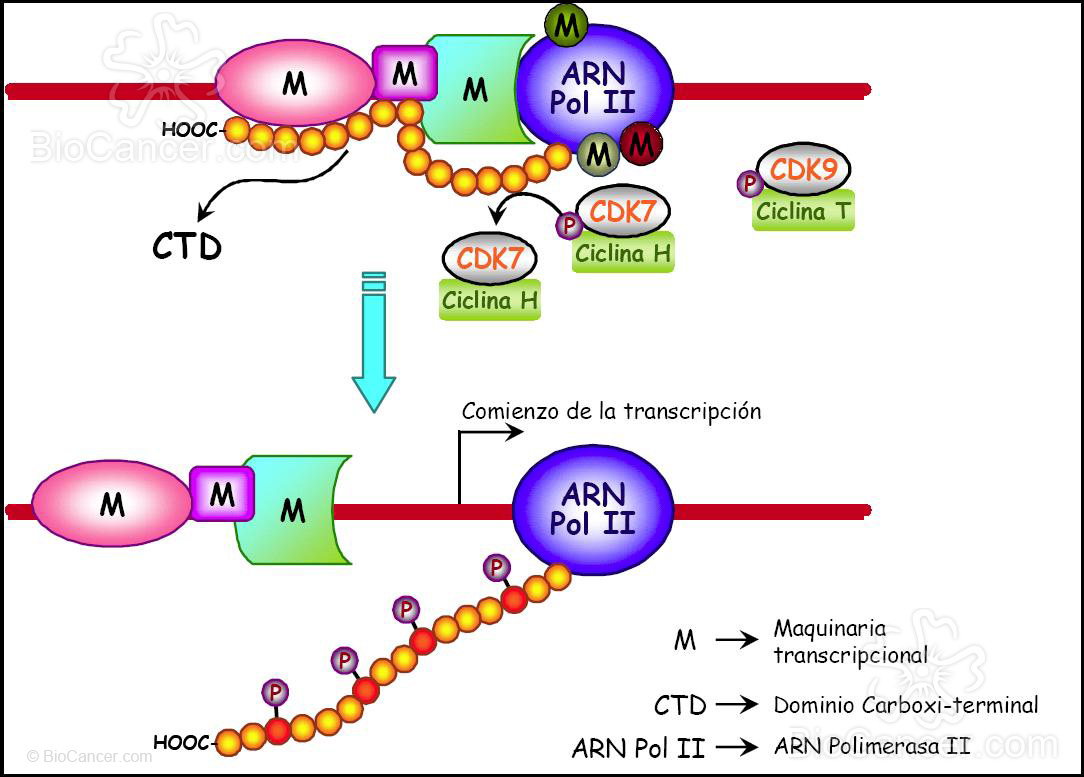
Figura 9. Una vez formada la maquinaria transcripcional, el complejo CDK7/ciclina H fosforila parcialmente el dominio carboxi-terminal de la ARN Polimerasa II, y promueve el inicio de la transcripción de la nueva molécula deA RN. Posteriormente, nuevas fosforilaciones llevadas a cabo por el complejo CDK9/ciclina T producen la elongación de la misma.
En vista de estos datos, los esfuerzos actuales se encaminan al diseño de pequeñas moléculas que sean capaces de ejercer una inhibición altamente selectiva sobre una o unas CDKs específicas frente a las otras.