3. La Ruta De Señalización Pi3k/akt/mtor
3. La Ruta De Señalización Pi3k/akt/mtor
La ruta de señalización de PI3K juega un papel fundamental en el metabolismo del cáncer (5), bien contribuyendo a la progresión del ciclo celular, bien disminuyendo la apoptosis e incrementando las capacidades metastásicas de las células cancerígenas (6,7). La activación descontrolada de la ruta de PI3K contribuye a la transformación celular y a la progresión tumoral en varios tipos de tumores incluyendo los de cerebro, mama, ovario y carcinomas renales. La activación de PI3K puede ocurrir a través de Ras activado o directamente por algunos receptores tirosina quinasa, que responden a varios factores de crecimiento y citoquinas como IL-1, IL-2, IL-3, IL-4, IL-6, factor de crecimiento tipo insulina (IGF), factor de crecimiento epidérmico (EGF), factor de crecimiento derivado de plaquetas (PDGF), factores de crecimiento de insulina (IGF-1 e IGF-2) y el factor estimulador de colonias (CSF). La activación de estos receptores tirosina quinasas conduce a la autofosforilación de la porción intracelular de los mismos, lo que sirve como punto de arranque para otras proteínas intracelulares.
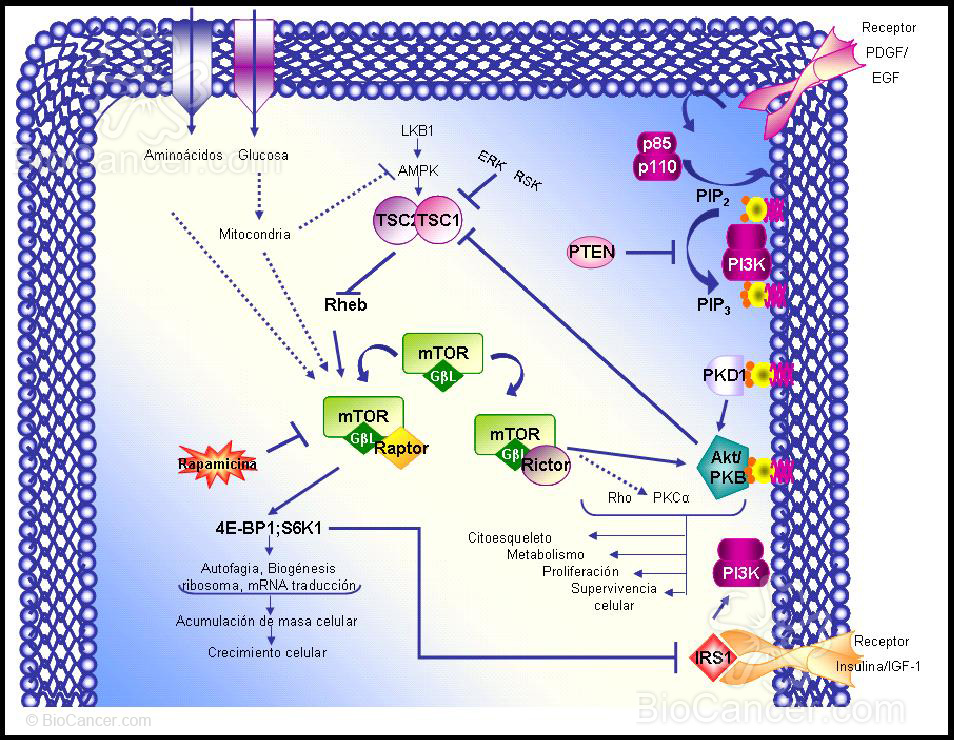
La ruta PI3K/Akt activada (Figura 1) se inicia a través del reclutamiento de PI3K a la membrana plasmática a través de la unión de su dominio SH2 a protefnas tirosina fosforiladas. En concreto, los residuos de tirosina fosforilados de los receptores tirosina quinasa interaccionan con la subunidad reguladora de PI3K, p85.
PI3K es un heterodfmero que consiste en una subunidad reguladora, p85, у una subunidad catalftica, pi 10, encargada de transferir el grupo fosfato у del ATP al fosfatidil inositol, 4,5-bifosfato (PIP2), generando el fosfatidil inositol, 3,4,5-trifosfato (PIPJ у ADP. Las actividades quinasas son reguladas por las fosfatasas que actúan eliminando los fosfatos de las protefnas diana. Existen evidencias de que PTEN desfosforila PIP3, actuando por tanto como regulador de la ruta de señalización de PI3K. PTEN tiene un dominio protefna-tirosina fosfatasa у un dominio de homologfa a la tensina, lo que sugiere que PTEN suprime el crecimiento celular tumoral ejerciendo un efecto antagonista al de las protefnas tirosina quinasas, regulando la invasión de las células tumorales у la metástasis a través de las interacciones con las adhesiones focales.
PIP3 sirve como ligando para reclutar la serina/treonina kinasa Akt (c-Akt, también llamada protefna-quinasa В, РКВ) a la membrana plasmática a través de la interacción directa con el dominio con homologfa plecstrina (PH) de Akt. Una vez en la cara interna de la membrana, Akt es fosforilado por una serina/treonina quinasa, la quinasa 1, dependiente de fosfatidil inositol-3 (PKD1), resultando en la activación de Akt (Figura 1).
La activación de Akt controla la supervivencia celular a través de la fosforilación de las dianas que dependen de ella, con el resultado neto del incremento en la supervivencia celular, proliferación, crecimiento у metabolismo. Las dianas para la activación de Akt pueden ser clasificadas en tres grupos distintos: protefnas apoptóticas, factores de transcripción у protefna-quinasas (Figura 2).
Akt fosforila directamente dos protefnas apoptóticas, BAD у caspasa 9, inhibiendo su actividad apoptótica у promoviendo por tanto la supervivencia celular (8) (Figura 2).
Los factores de transcripción pueden bien ser activados 0 inhibidos tras la fosforilación de Akt. Akt activa el factor de transcripción NF-kB, HIF-la у CREB, lo que tiene como consecuencia un incremento en la transcripción de genes anti-apoptóticos (Figura 2).
El factor de transcripción NF-kB es el mediador central de la respuesta inmune, de la respuesta inflamatoria у la respuesta de supervivencia celular(9). NF-kB es activado por Akt a través de la fosforilación de la quinasa inhibidora de U?-VB(IKKap). Tras su activación, IKK fosforila IkB, marcándolo para la ubiquitinación у degradación en el proteosoma. Esto expone los lugares de localización nuclear de NF-kB у le permite la traslocación al núcleo donde induce la expresión de genes anti-apoptóticos (8, 10). Los factores de crecimiento como EGF activan NF-kB у protegen contra la apoptosis y, por el contrario, la inhibición de NF-kB sensibiliza a la célula a una amplia variedad de estfmulos pro-apoptóticos (11) (Figura 2).
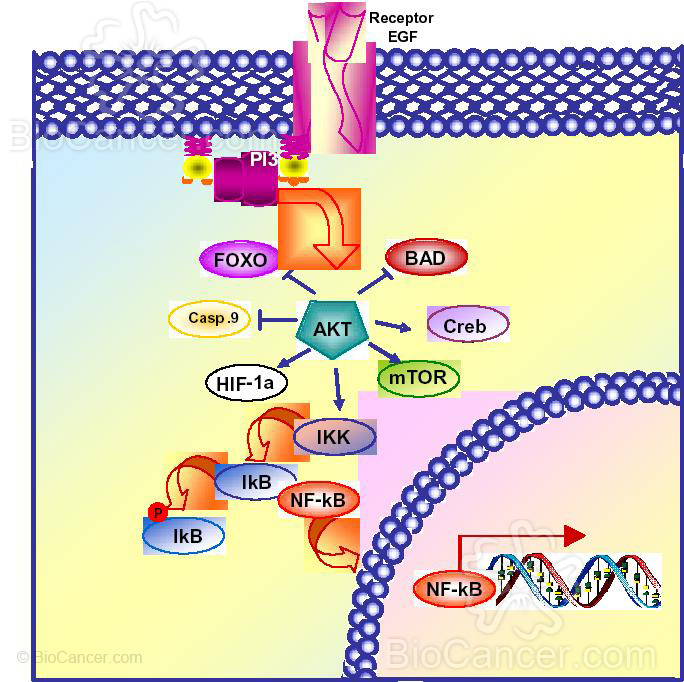
Además, la activación de HIF-1 inducida por EGF y mediada por Akt conduce a una mayor expresión del factor de crecimiento endotelial (VEGF) que protege a las células de la apoptosis. Akt también fosforila directamente CREB activando su actividad transcripcional y sobreexpresando genes antiapoptóticos como Mcl-1 (12). Por el contrario, Akt inactiva factores de transcripción FOXO (familia Forkhead) y p53, bien directamente fosforilando proteínas FOXO o por fosforilación y activación de MDM2, regulador negativo de p53 (13, 14). En ambos casos, la expresión de genes proapoptóticos disminuyen causando un aumento de la supervivencia celular.
Gsk-3 también es una diana de fosforilación de Akt, lo que determina su inactividad, bloqueando su actividad transcripcional y la regulación de su metabolismo. La inhibición de Gsk-3 protege a las células de la apoptosis pero el mecanismo exacto no se conoce.
Específicamente, la fosforilación de mTOR por Akt ocurre a través de la inactivación del complejo de esclerosis tuberosa (TSC). El complejo TSC es un heterodímero que consiste en tuberina (TSC2) no fosforilada y hamartina (TSC1), que actúa como proteína GTPasa activadora (GAP), inhibiendo a la pequeña proteína G-Rheb. Por fosforilación de TSC2, Akt interrumpe el complejo, permitiendo a Rheb, unirse a ATP y pasar desde el estado GDP inactivo al estado activo. Rheb, unido a GTP, activa a mTOR (15) (Figura 1).
En una ruta paralela la proteína AMPK fosforila, aunque en este caso activa, TSC2, inhibiendo así la activación de mTOR en respuesta a cambios intracelulares de la relación ATP/AMP. Esto une la ruta que detecta niveles de energía (a través de aminoácidos y ATP), con la ruta de mTOR. LKB1, una serina-treonina quinasa (también llamada STK11), es una quinasa activadora de AMPK, que sirve como un importante inhibidor de la ruta mTOR en respuesta a la escasez de energía (16) (Figura 1).